平山病诊断、鉴别
更新日期:2019-10-18 02:34:40 来源:本站作者:admin平山病临床易与肌萎缩侧索硬化或脊髓进行性肌萎缩等运动神经元病混淆,但绝大多数患者在以后5年内病情可自然中止,预后与运动神经元病明显不同。
1、平山病诊断依据:
(1)平山病临床表现主要有:①青春期早期隐匿起病,男性多见;②局限性上肢远端、手指及腕无力,伴有手及前臂远端肌群萎缩;③寒冷麻痹和手指伸展时束颤;④症状为单侧或以一侧明显;⑤无感觉异常,颅神经损害,毒物暴露史,下肢、括约肌、脑干损害;⑥发病后数年间呈进行性(85%患者发病后1-5年停止进展)。
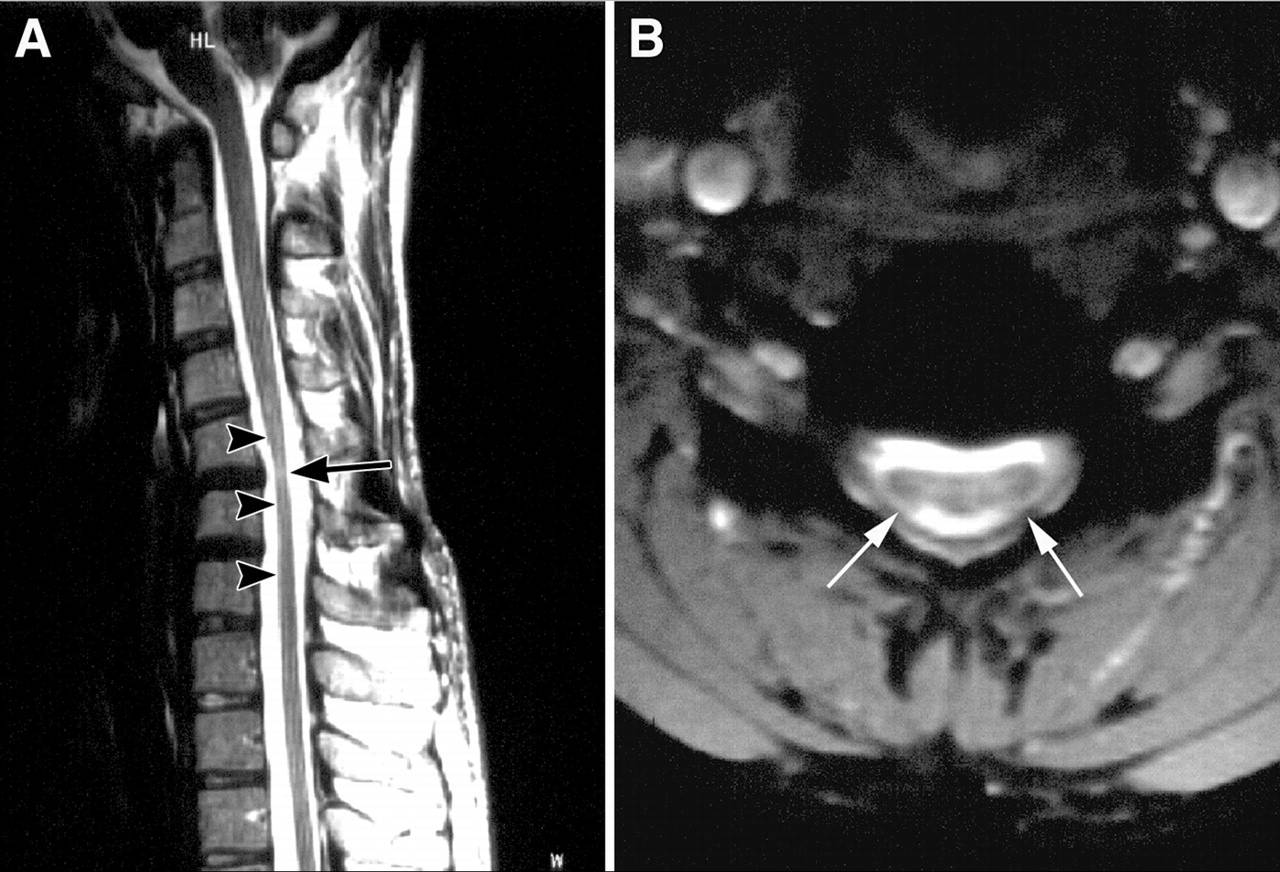
(2)辅助检查:①电生理:肌电图检查示萎缩肌肉呈神经源性损害,对侧无萎缩的同名肌肉也可见神经源性损害,但周围神经传导速度正常。②影像学检查:自然状态下MRI可见脊髓非对称性前角萎缩和硬膜外静脉丛扩张;曲颈状态下(平卧位曲颈下颌靠近胸部)MRI检查可见颈髓的硬脊膜后壁向前推移使下部颈髓受压,脊膜后有月牙形异常信号影。
2、平山病诊断标准:
(1)肯定:具备“临床表现”和“辅助检查”各项;
(2)可能:缺少“临床表现”中除第六项以外的一项,但具备“辅助检查”各项;
(3)可疑:缺少“临床表现”中除第六项以外两项以上,但具备“辅助检查”各项,并排除其他可能相关疾病。平山慧造认为本病诊断的主要依据为临床特征,病程为自限性尤为重要,肌电图检查及MRI发现椎管后壁前移压迫脊髓对诊断有重要意义。
3、平山病鉴别诊断:
(1)进行性脊肌萎缩症:首发症状常为一只手或双手小肌肉萎缩、无力,渐累及前臂、上臂,远端萎缩明显,肌无力,腱反射降低,无感觉障碍及括约肌障碍,与平山病临床上相似,但进行性脊肌萎缩症的起病年龄多在30岁以后,病程呈进展性,至少有两个部位受损,可波及延髓,磁共振成像显示受累脊髓和脑干萎缩变小,但平山病无此表现。
(2)多灶性运动神经病:为非对称性的肢体远端的肌无力伴有肌萎缩,以上肢为主,无感觉及括约肌障碍,无锥体束征,与平山病相似,但多灶性运动神经病发病年龄多在40岁以上,病程呈缓慢进行性,肌电图特点是运动神经出现特征性多灶性传导阻滞。
(3)慢性运动轴索性神经病:是最近报道的一种周围神经病,临床主要表现为进行性肌萎缩、无力,感觉正常,但其电生理特点是复合肌肉运动电位波幅普遍性降低,可鉴别。
(4)脊髓萎缩症:尤其是脊髓萎缩症III型即青少年型,表现为下运动神经元损伤,肌萎缩无力,但多从四肢近端开始,病程呈进展性,为常染色体隐性遗传,基因检测可以鉴别。
(5)普通脊髓型颈椎病:由于支配颈、胸、腰、骶的神经纤维在皮质脊髓束中从内到外的排列顺序,所以脊髓受压后运动障碍是先下肢后上肢,表现为步态笨拙、摇摆不稳、下肢无力、肌张力增高和出现病理反射,早期可出现动态Hoffmann征(即患者在头颈部后伸状态下引出该反射),并多数有颈肩部麻痹酸痛等症状,可以与本病相鉴别。
如果您还有什么不明白的,请咨询在线医生或拨打咨询热线400-7078-333。



























 冀公网安备 13010402001899号
冀公网安备 13010402001899号